Die Mastozytose kann sich durch viele verschiedene unspezifische Symptome äußern. Zudem kommen verschiedene Formen der Mastozytose vor. Bei der kutanen Mastozytose entstehen vor allem Veränderungen auf der Haut, bei der systemischen Mastozytose ist der ganze Körper betroffen. In diesem Beitrag erfahren Sie, welche Formen es gibt und welche Symptome auftreten können.
Welche Formen von Mastozytose gibt es?
Die Mastozytose wird in verschiedene Formen eingeteilt. Zunächst in die Mastozytose der Haut (kutane Mastozytose) und die Mastozytose des gesamten Körpers (systemische Mastozytose). Diese beiden Formen werden weiter unterteilt, je nachdem, wo im Körper die Mastzellen vermehrt auftreten und welche Beschwerden sie verursachen.
Kutane Mastozytose (CM)
Hautmastozytosen werden auch kutane Mastozytosen (kutan = die Haut betreffend) genannt. Hier vermehren sich die Mastzellen in der Regel nur in der Haut. Hautmastozytosen treten meistens bei Kindern auf und sind gutartig. Bei vielen heilen sie im Verlauf der Pubertät ganz aus. Wenn Erwachsene eine Hautmastozytose haben, können auch zusätzlich Symptome an anderen Stellen im Körper auftreten.
Die Hautmastozytose wird in verschiedene Formen unterteilt:
Makulopapulöse kutane Mastozytose (MPCM)
Sie ist die häufigste Form der Hautmastozytose. Die MPCM zeichnet sich durch kleine braune bis rotbraune Flecken auf der Haut aus, meist am Oberkörper und auf den Oberschenkeln. Das Gesicht bleibt normalerweise ausgespart.
Telangiectasia macularis eruptiva perstans (TMEP)
Diese seltene Unterform Form MPCM tritt meist bei Erwachsenen auf. Betroffene haben kleine rosafarbene Hautflecken, die von erweiterten Blutgefäßen durchzogen sind.
Diffuse kutane Mastozytose (DCM)
Die DCM ist eine sehr seltene Form, die vor allem bei Babys und kleinen Kindern unter drei Jahren auftritt. Durch besonders viele Mastzellen in der Haut entstehen teigige Verdickungen oder Verhärtungen, die leicht rot, gelb oder braun verfärbt sein können. Häufig bilden sich Blasen.
Mastozytom
Ein Mastozytom ist selten und kommt meistens bei Kindern vor. Es erscheint in der Regel als großer, dicker rötlich-gelblich-brauner Fleck. Selten tritt mehr als ein Mastozytom auf.
Hinweis: Eltern und Angehörigen von Kindern mit einer Mastozytose steht das Kindernetzwerk Mastozytose zur Seite.
Systemische Mastozytose (SM)
Häufen sich Mastzellen in inneren Organen an, beispielsweise im Knochenmark, in der Leber oder der Milz sprechen Fachleute von einer systemischen Mastozytose. Auch die Haut kann zusätzlich betroffen sein. Meist tritt die systemische Mastozytose bei Erwachsenen auf. Sie ist nicht heilbar, viele Menschen können aber gut damit leben, wenn sie erkannt haben, worauf sie achten müssen.
Die systemische Mastozytose wird in verschiedene Formen unterteilt:
(Indolente) systemische Mastozytose (ISM)
Die (indolente) systemische Mastozytose ist die häufigste Form. Sie betrifft etwa die Hälfte aller Patientinnen und Patienten mit einer systemischen Mastozytose. Sie verläuft meist langsam fortschreitend und Betroffene haben in der Regel eine normale Lebenserwartung. Der Begriff „indolent“ bedeutet „schmerzabgewandt“. Diese Bezeichnung sollte aber nicht zu der Schlussfolgerung führen, dass sich eine indolente Mastozytose kaum auf das Leben Betroffener auswirkt. Auch bei Menschen, die an einer indolenten Form erkrankt sind, kann die Lebensqualität mitunter sehr stark beeinträchtigt sein.
Smouldering Systemische Mastozytose (SSM)
Eine seltene Unterform der (indolenten) systemischen Mastozytose ist die „schwelende“ Variante, in der auch das Knochenmark von einer Mastzellansammlung betroffen und Organe krankhaft vergrößert sein können. Sie ist deshalb etwas aggressiver als die (indolente) systemische Form.
Fortgeschrittene systemische Mastozytose (ADvSM)
Bei der fortgeschrittenen Mastozytose (AdvSM, abgekürzt aus dem Englischen für Advanced Systemic Mastocytosis) haben die Mastzellen Leber, Milz, Darmtrakt, Lymphknoten oder Knochenmark besiedelt. Dadurch funktionieren die betroffenen Organe nicht mehr richtig. Die fortgeschrittene systemische Mastozytose wird in drei Unterformen unterteilt:
- Systemische Mastozytose mit begleitender Blutbildungserkrankung (SM-AHN)
Bei dieser Form liegt zusätzlich eine weitere Knochenmarkerkrankung vor. Sie ist die häufigste Variante der fortgeschrittenen systemischen Mastozytose.
- Aggressive systemische Mastozytose (ASM)
Bei der aggressiven systemischen Mastozytose haben sich die Mastzellen in den Organen so stark vermehrt, dass sie die jeweiligen Organe schädigen. Die Organe vergrößern sich und funktionieren nicht mehr richtig. Die Haut ist meistens nicht betroffen.
- Mastzellleukämie (MCL)
Extrem selten entarten die Mastzellen so, dass es zu einer Mastzellleukämie (Blutkrebs) kommt. Diese ist sehr gefährlich.
Welche Symptome verursacht eine Mastozytose?
Menschen mit Mastozytose werden häufig erst sehr spät diagnostiziert, weil es wenig „typische“ Anzeichen gibt. Es kann zu Beschwerden im ganzen Körper kommen.
Die häufigsten Symptome beschreiben wir im folgenden Abschnitt.
Symptome auf der Haut
8 bis 9 von 10 Menschen mit Mastozytose zeigen Symptome auf der Haut. Das heißt nicht zwingend, dass es sich um eine kutane Mastozytose handelt. Auch bei einer systemischen Mastozytose leiden die meisten Betroffenen zusätzlich unter Hautveränderungen. Häufig sind Symptome auf der Haut dann das erste Anzeichen, das Ärztinnen und Ärzte auf die richtige Spur führt. Folgende Symptome treten häufig auf:
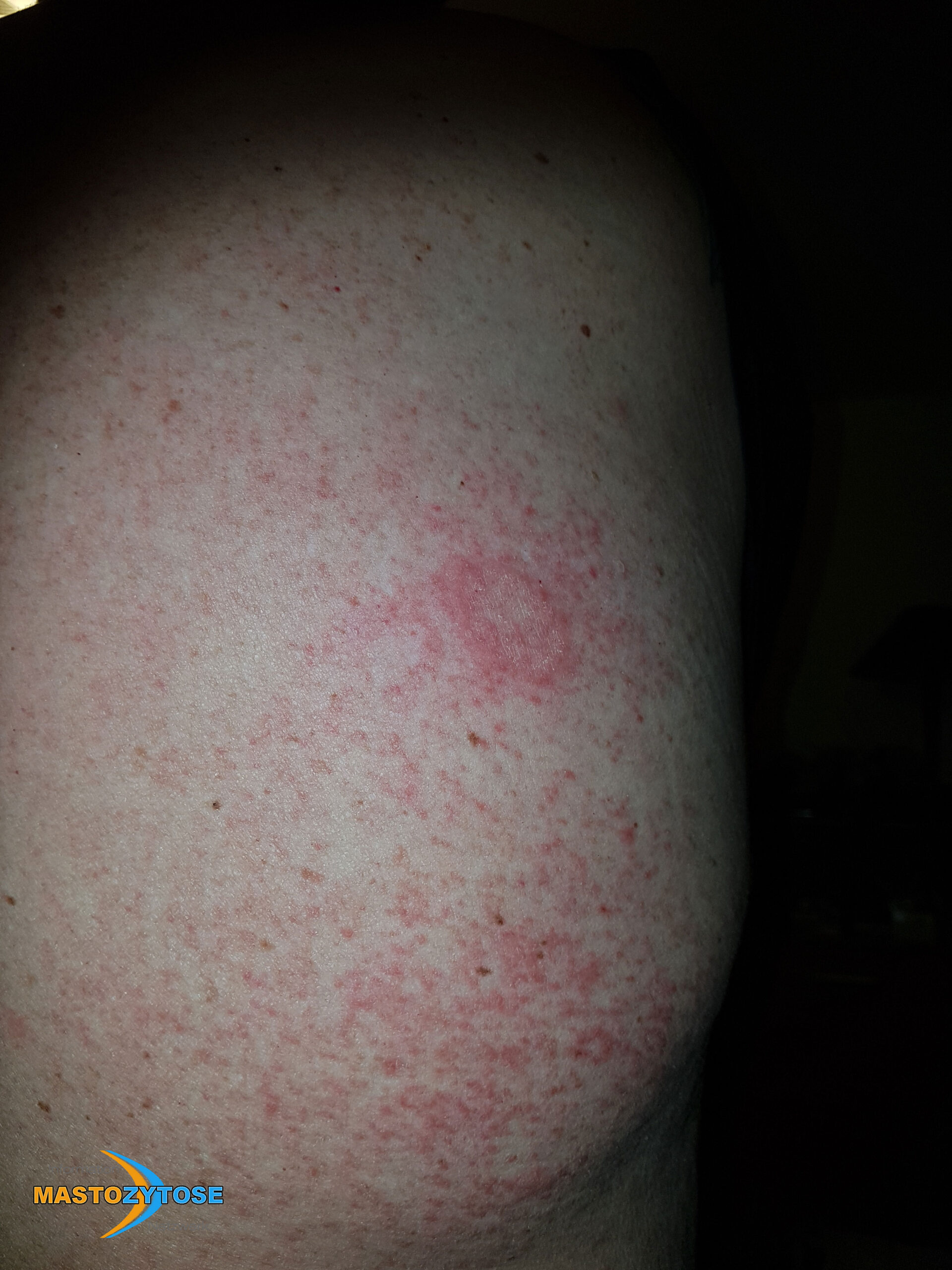
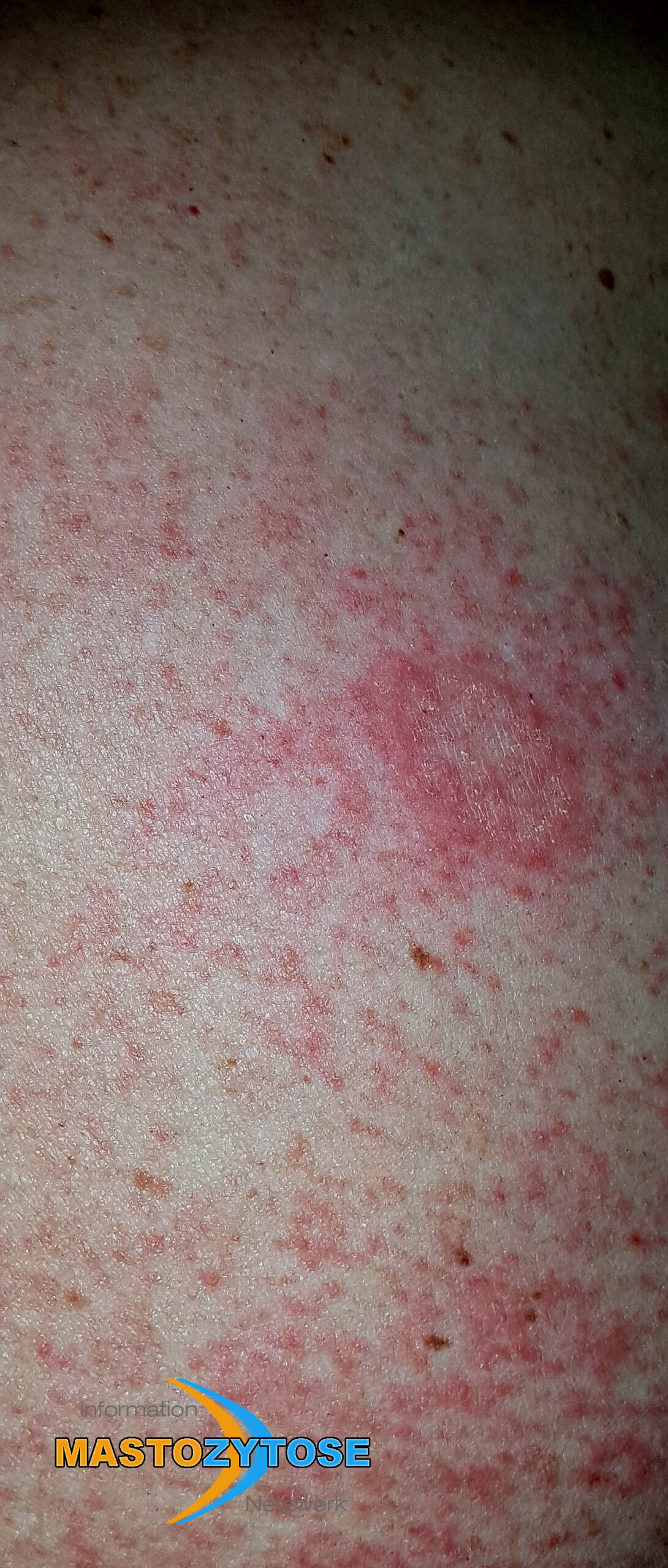
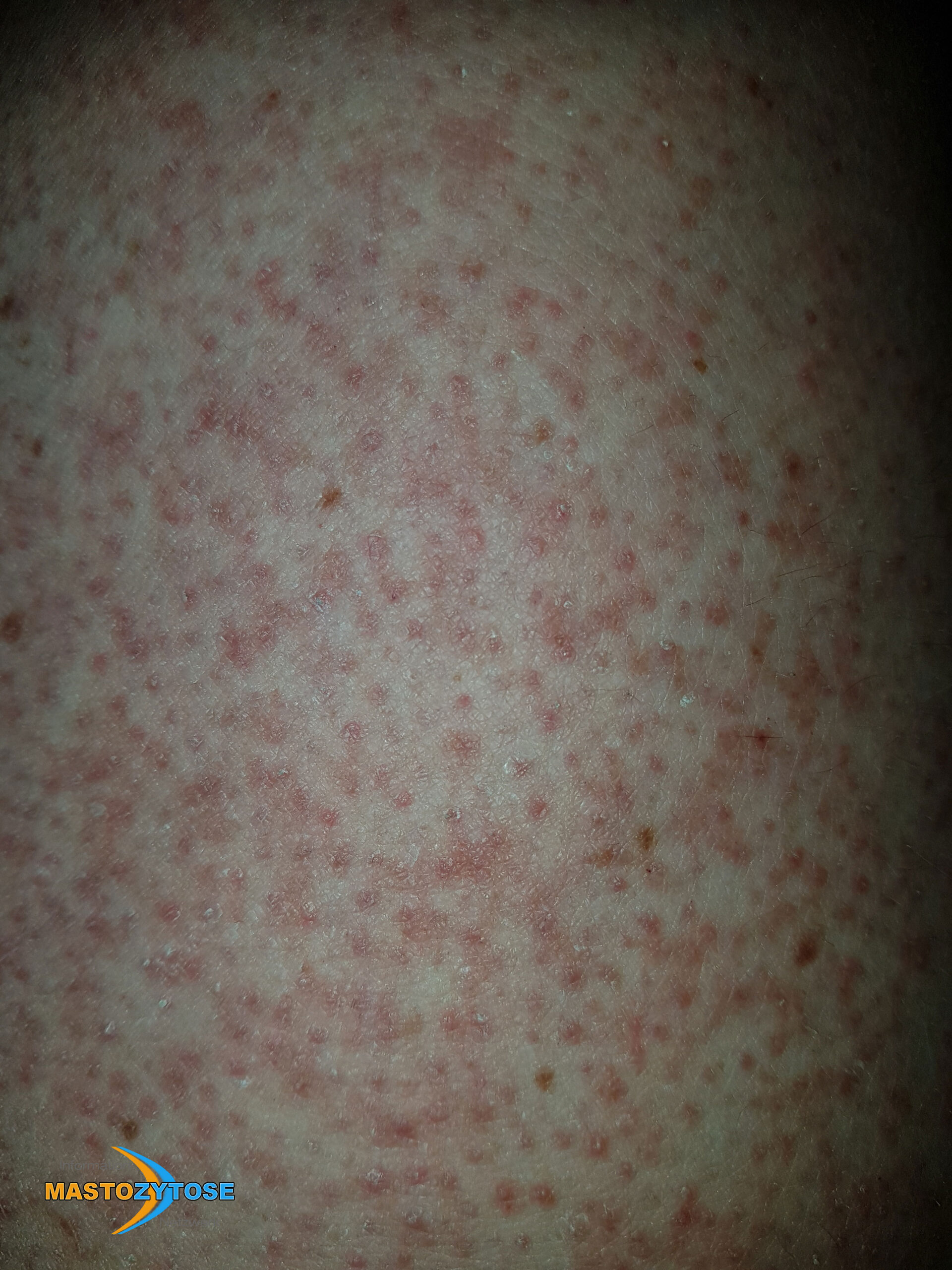
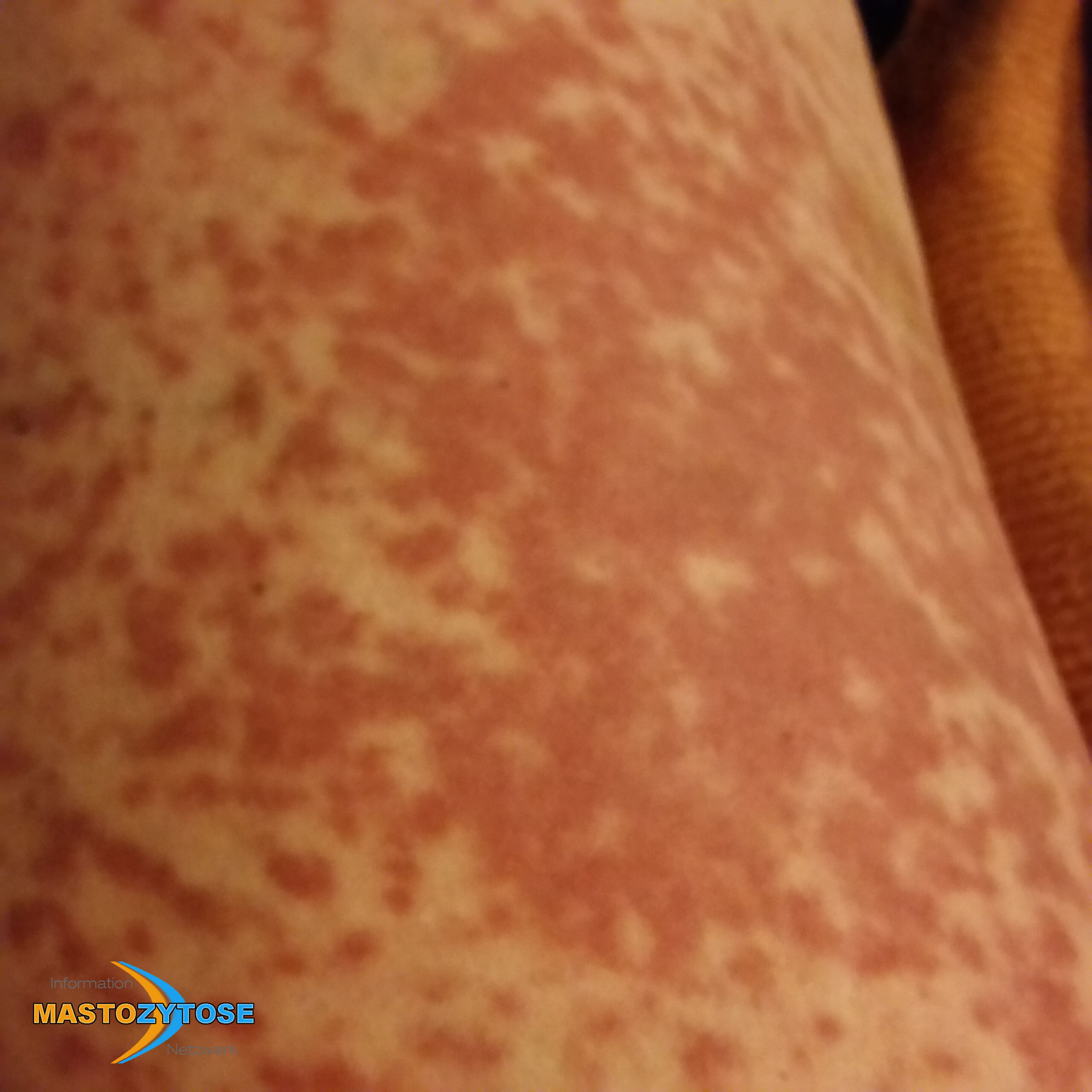
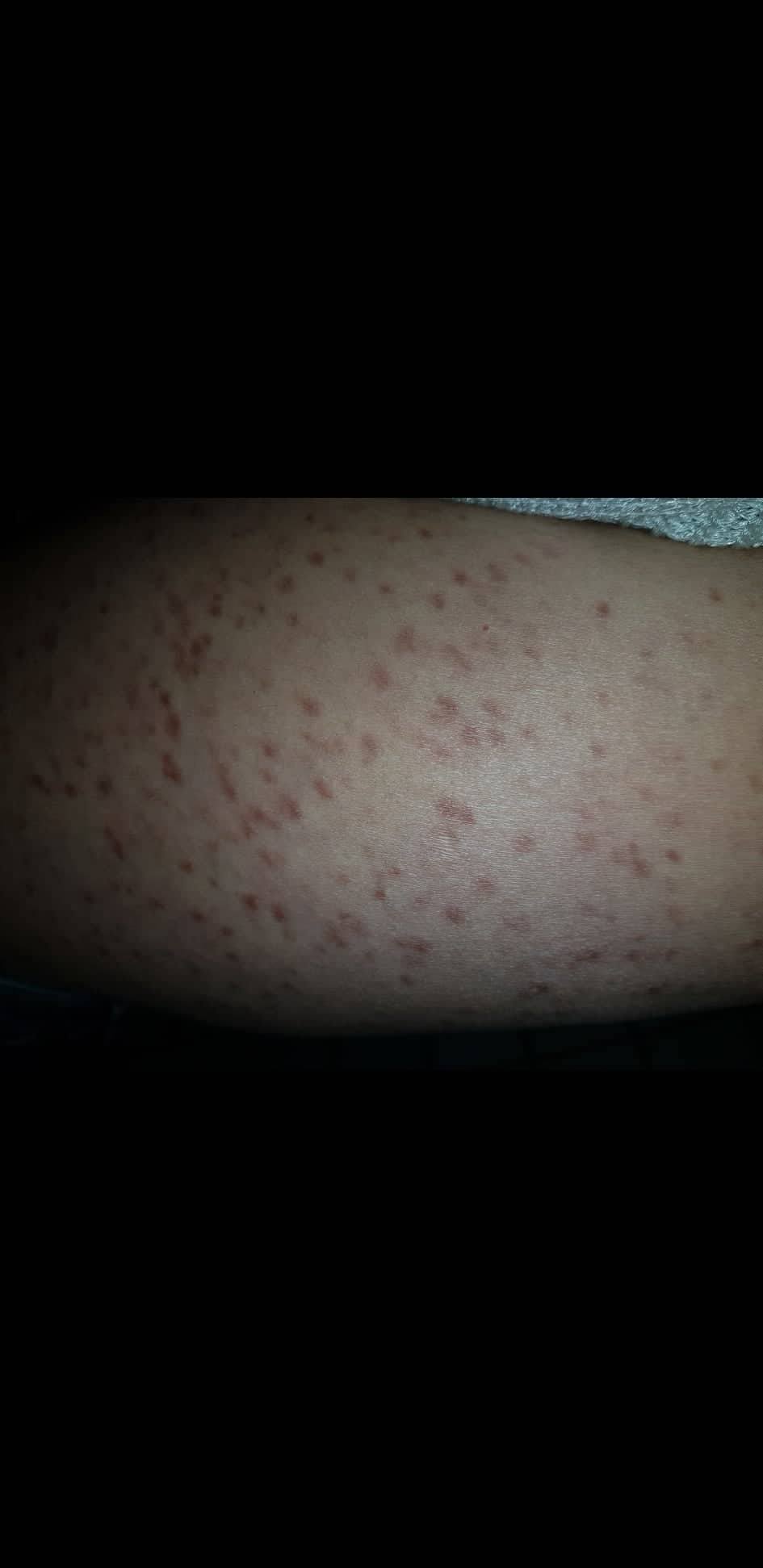
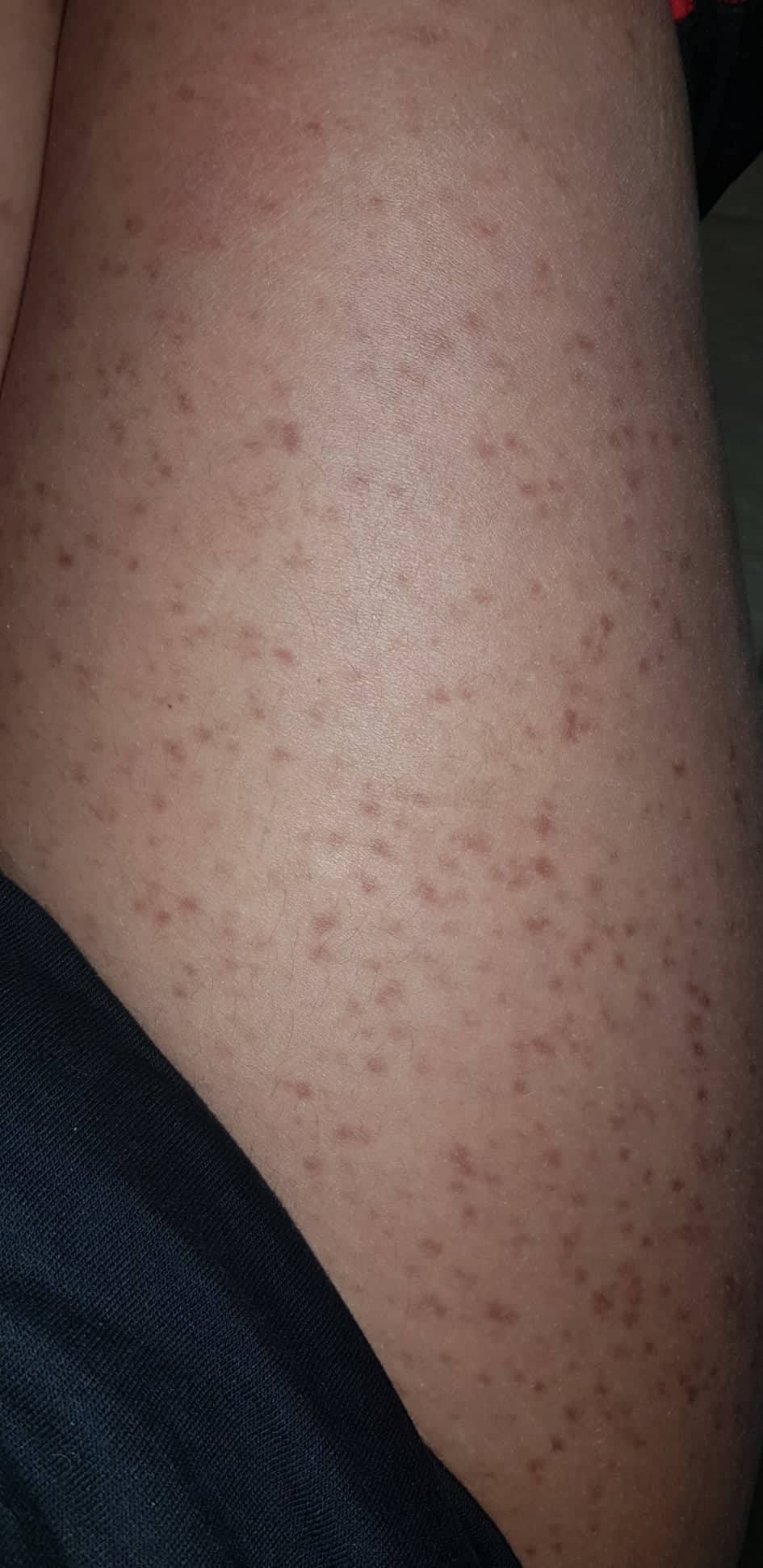
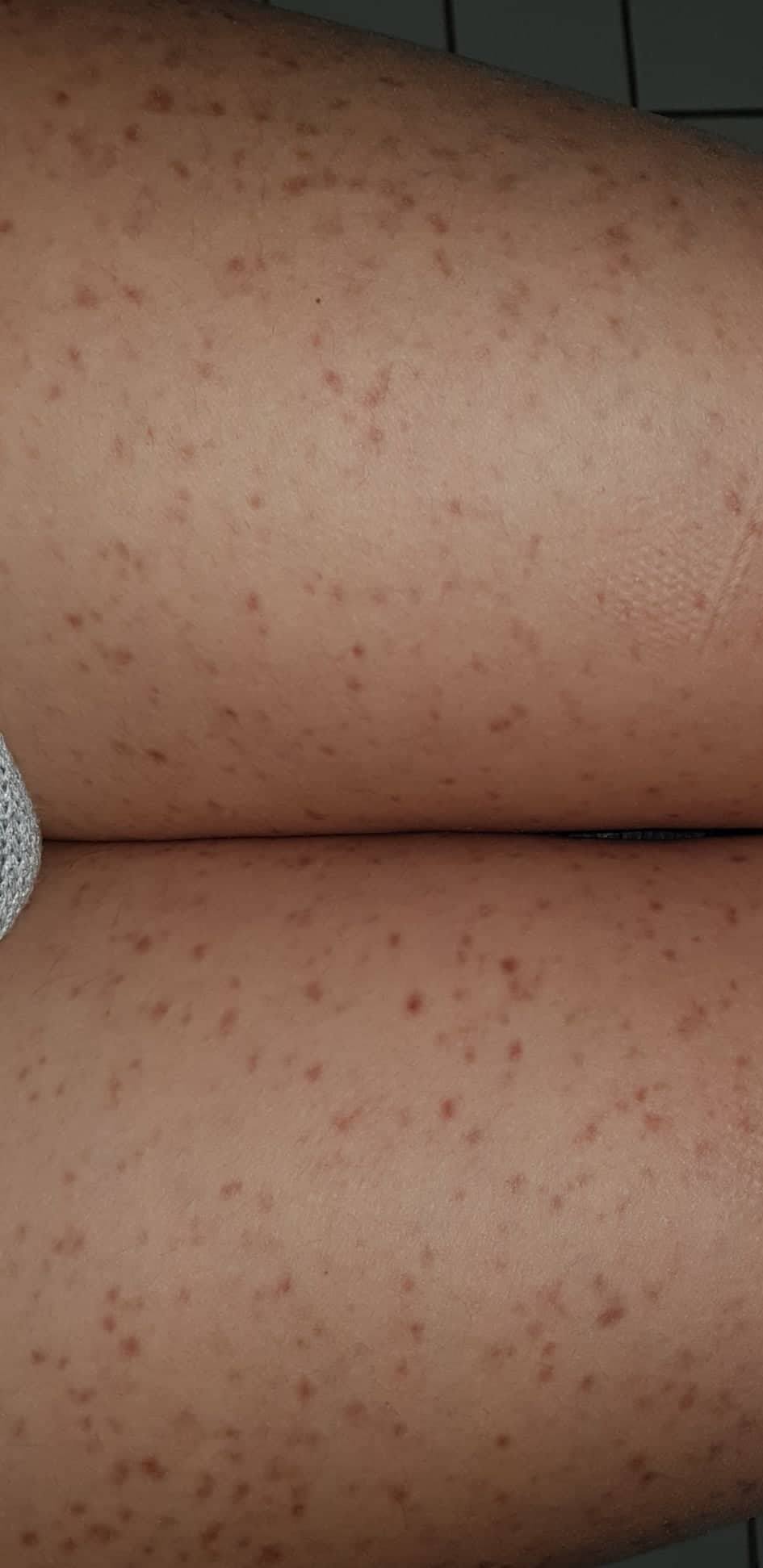
- Flecken: Häufig entstehen rotbraune oder gelbliche Flecken, meistens auf den Oberschenkeln und am Oberkörper. Sie können sich auch mit der Zeit auf den Rest des Körpers ausweiten. Kopf, Hand- und Fußflächen sind in der Regel nicht betroffen.
- Quaddeln oder Blasen: Durch Druck und Reibung schwellen die Flecken oft an und werden zu juckenden Quaddeln.
- Rötungen und Hitzewallungen: Manche Betroffene bekommen regelmäßig plötzliche Rötungen der Haut in Kombination mit einer Hitzewallung. Dies wird auch Flush genannt.
Bilder von Betroffenen
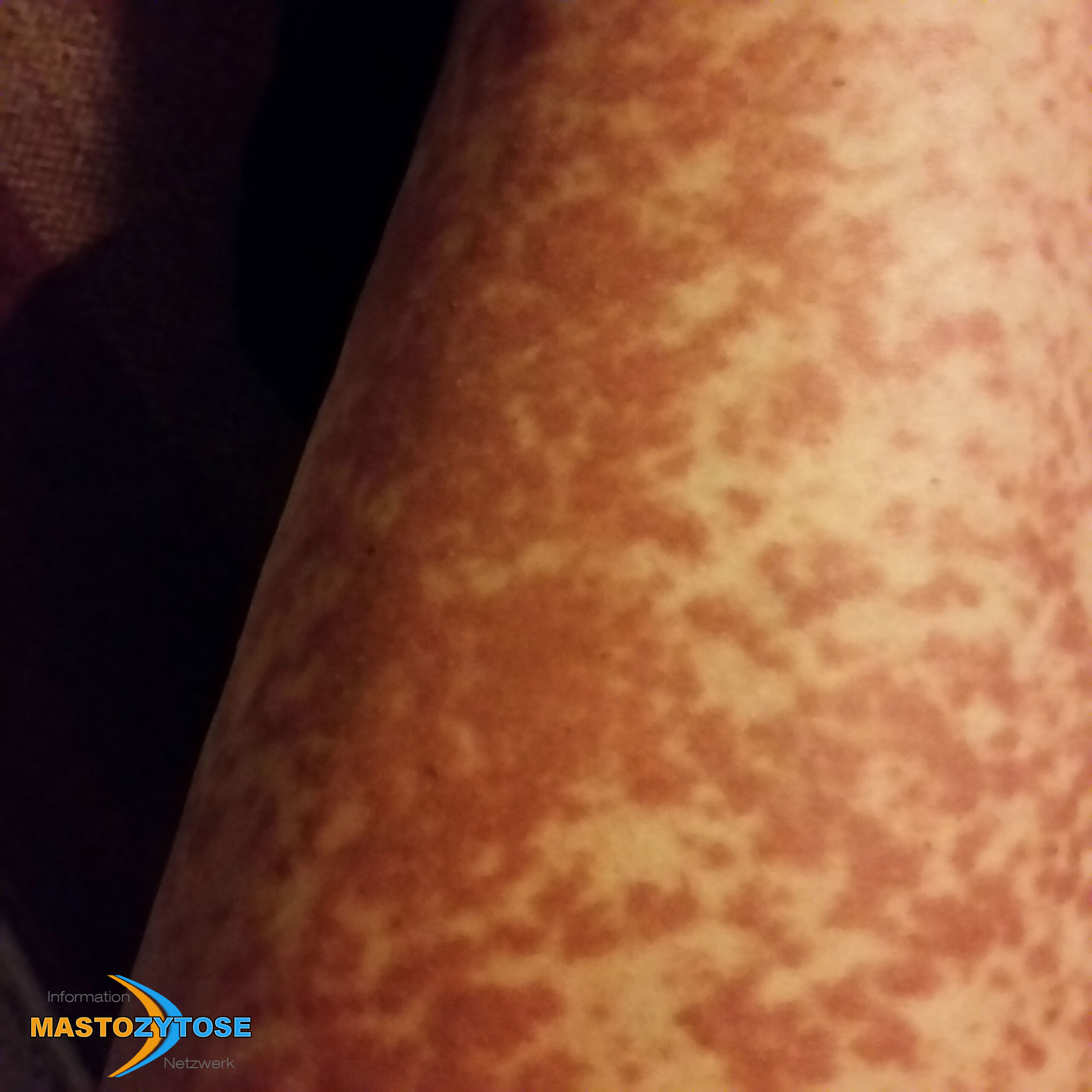
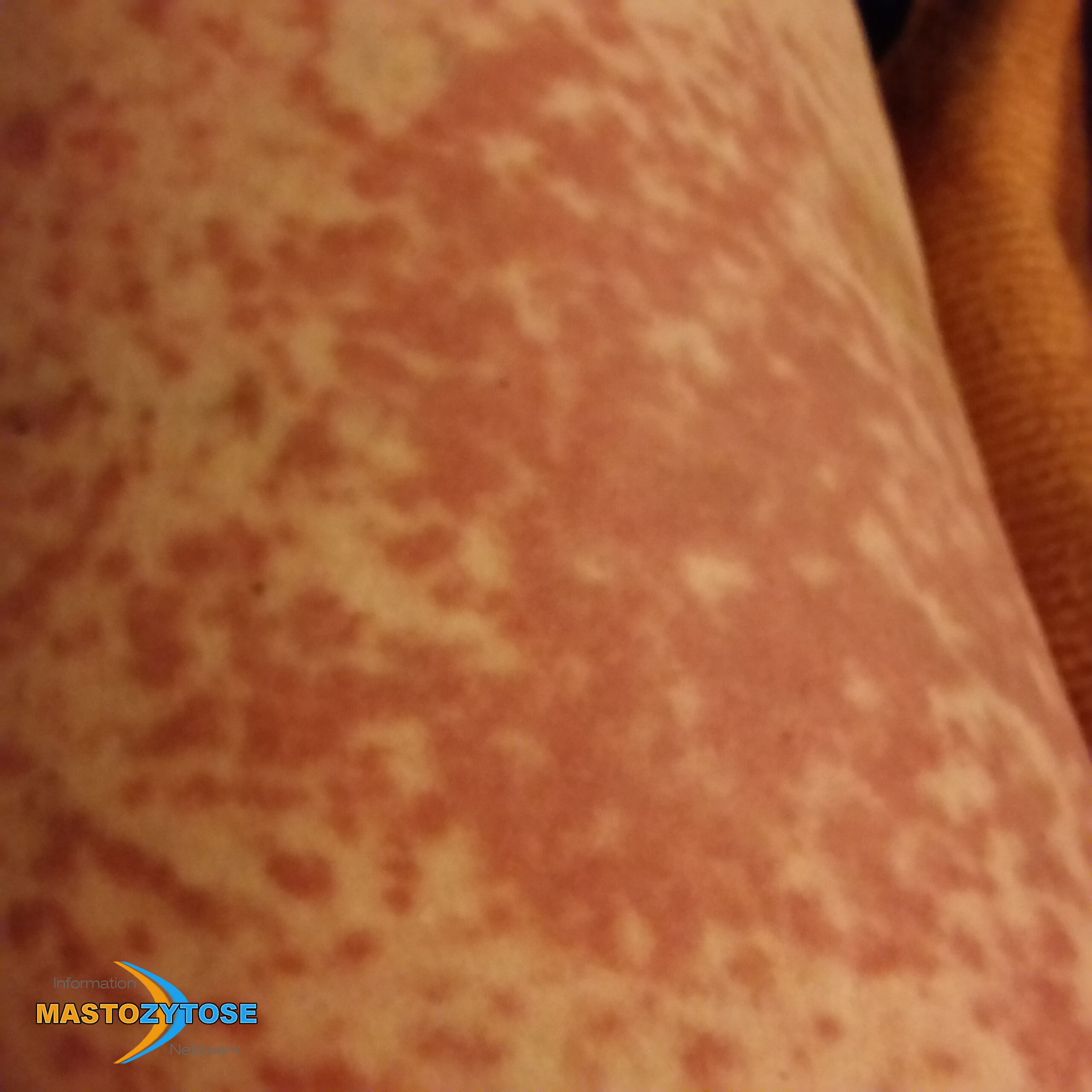
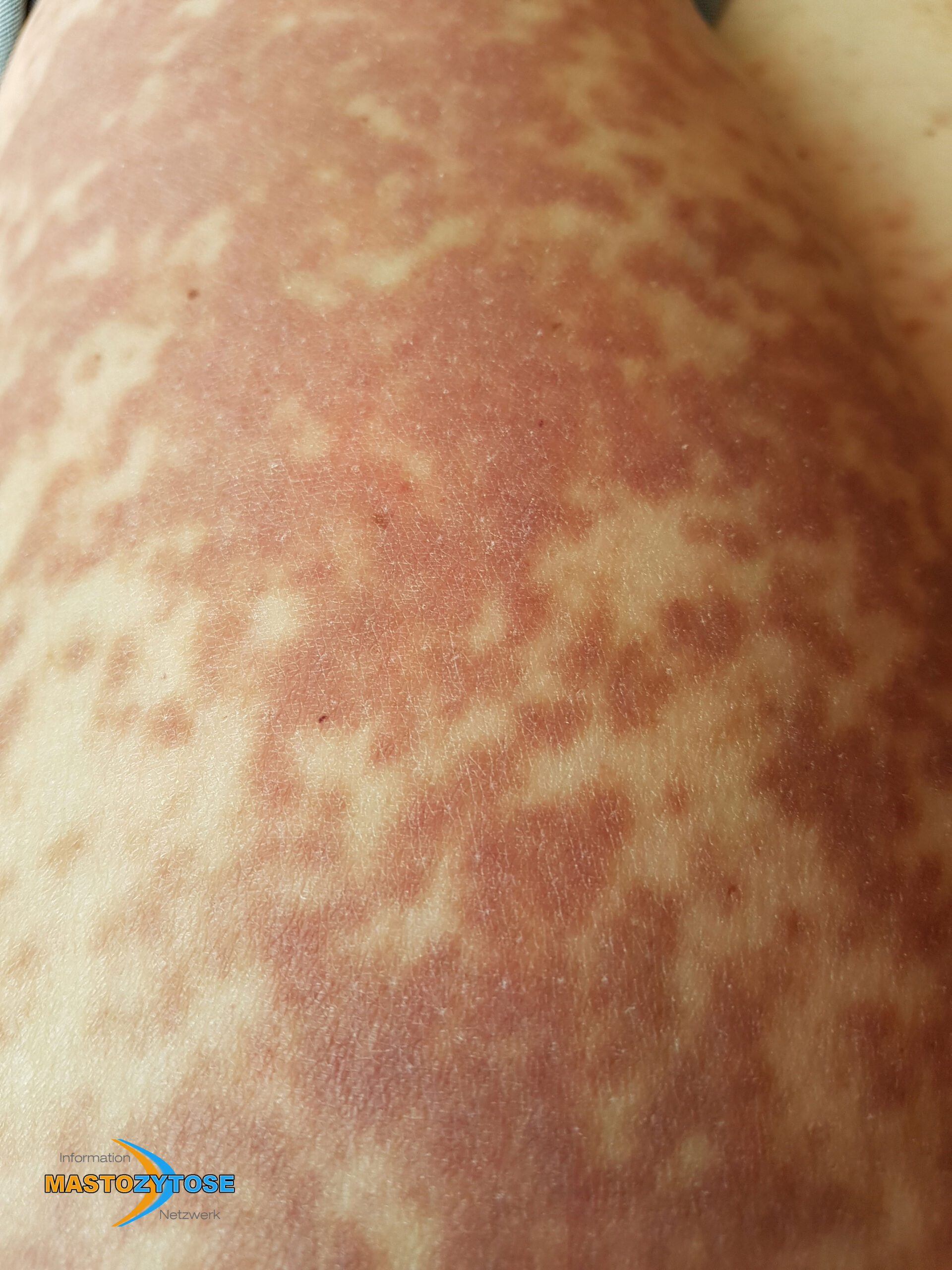
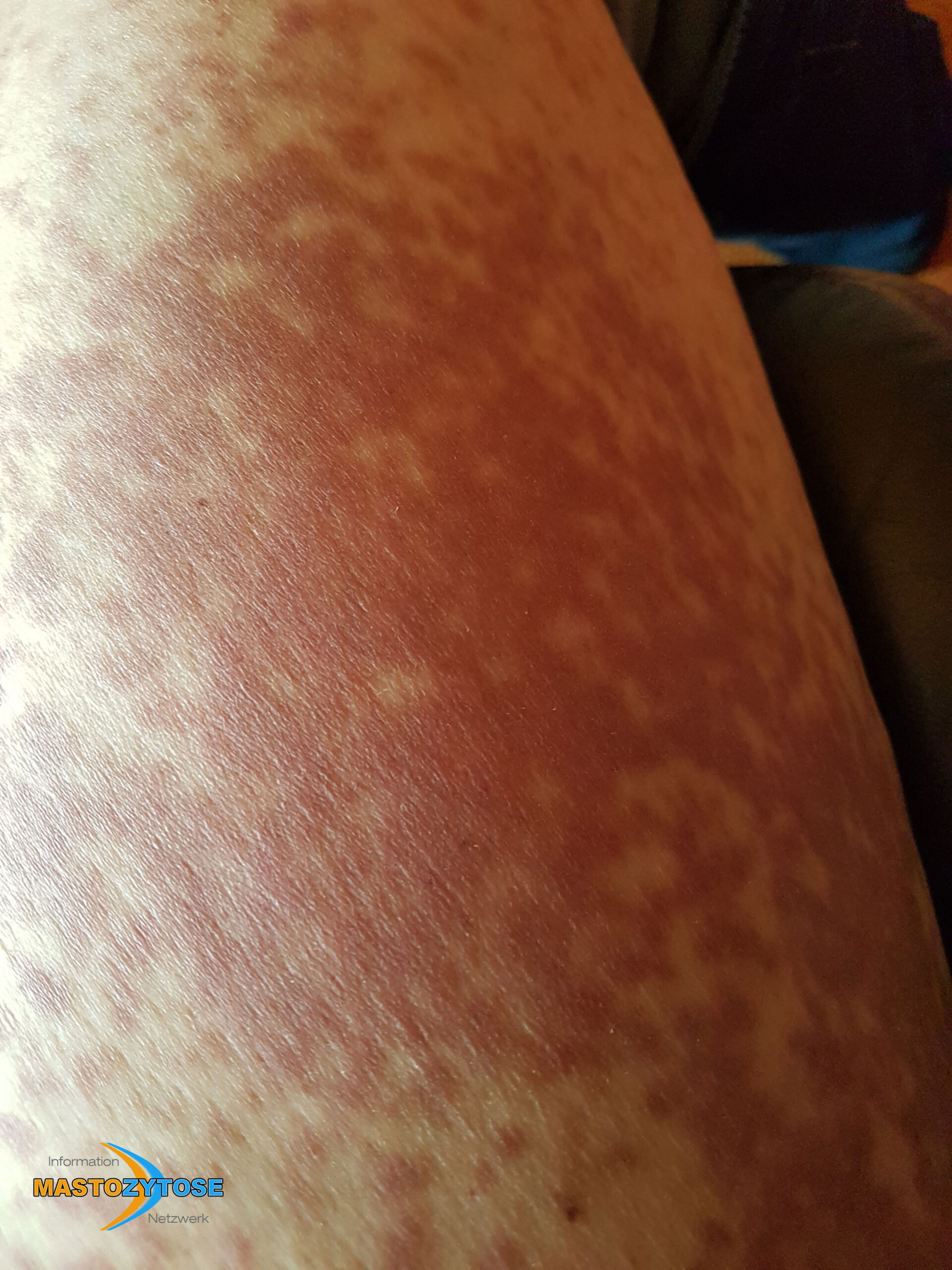
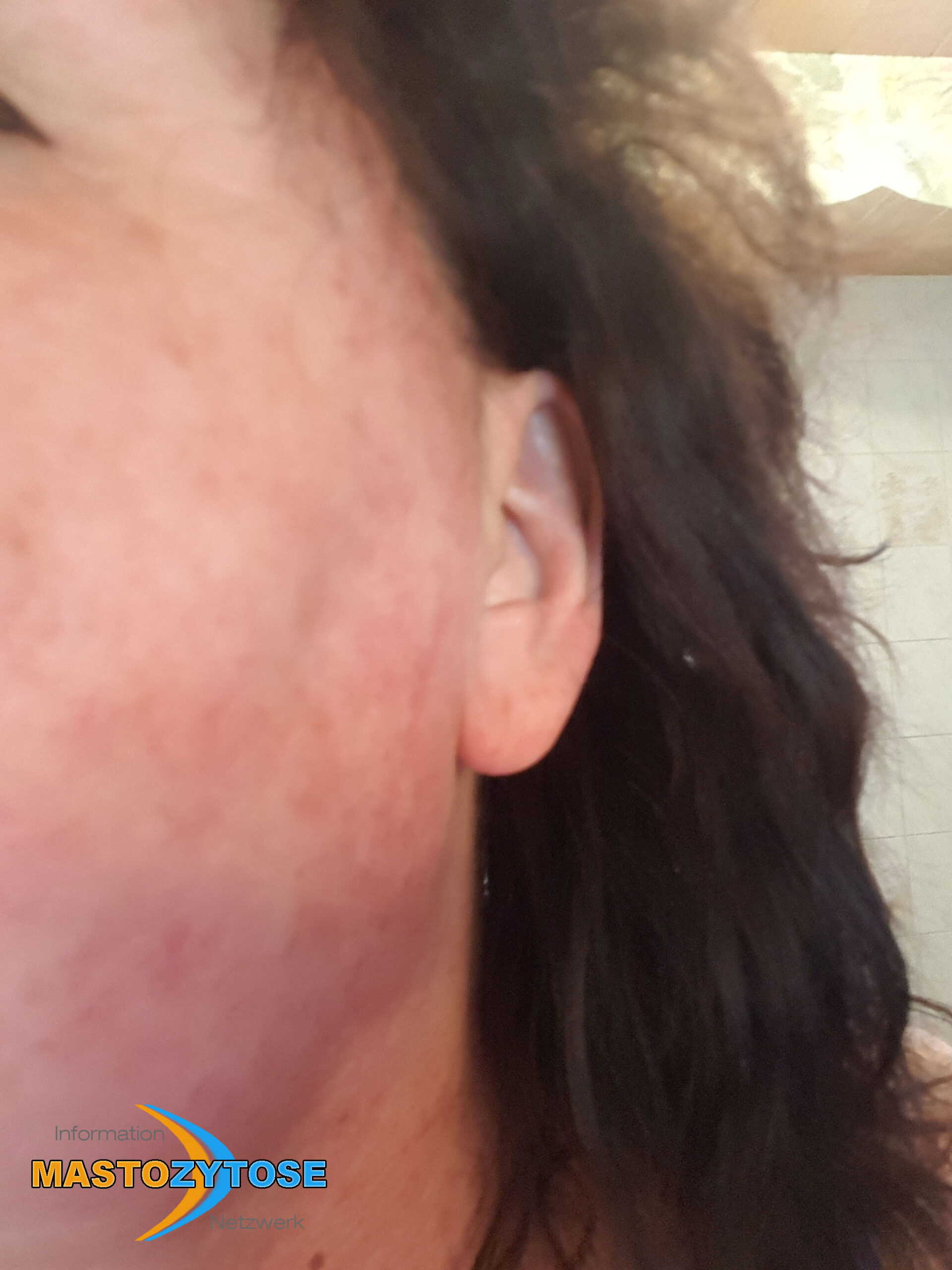
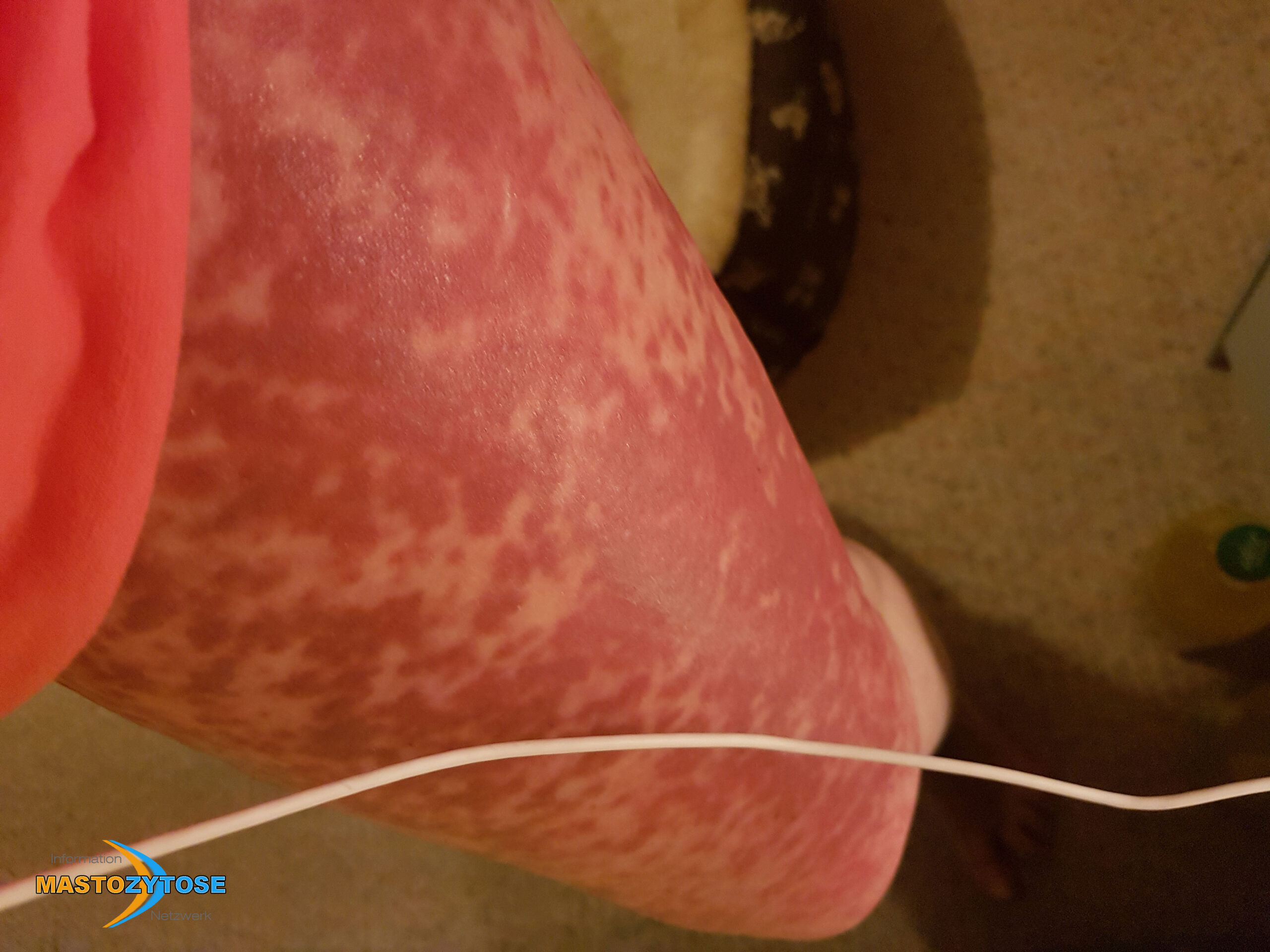
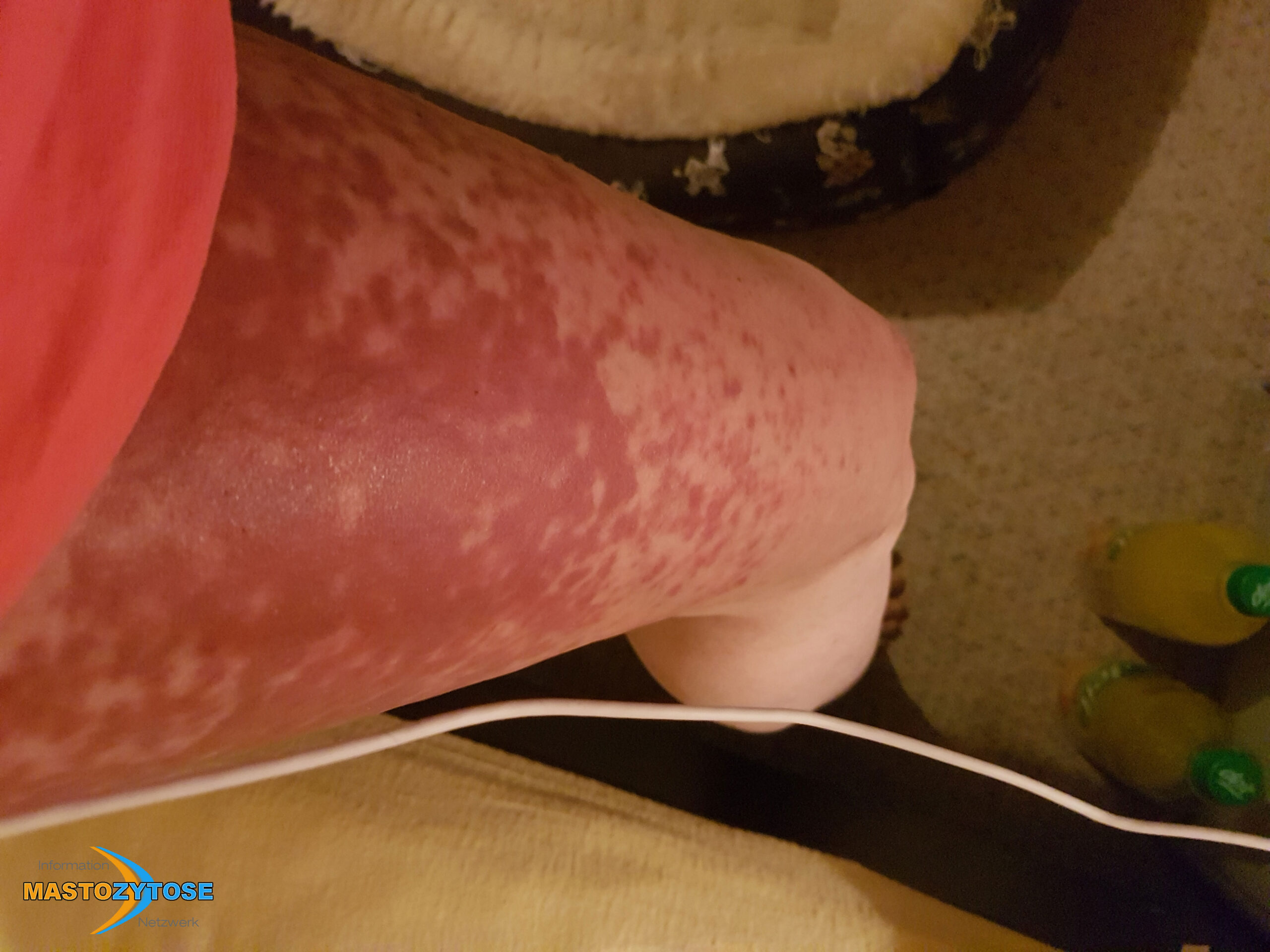
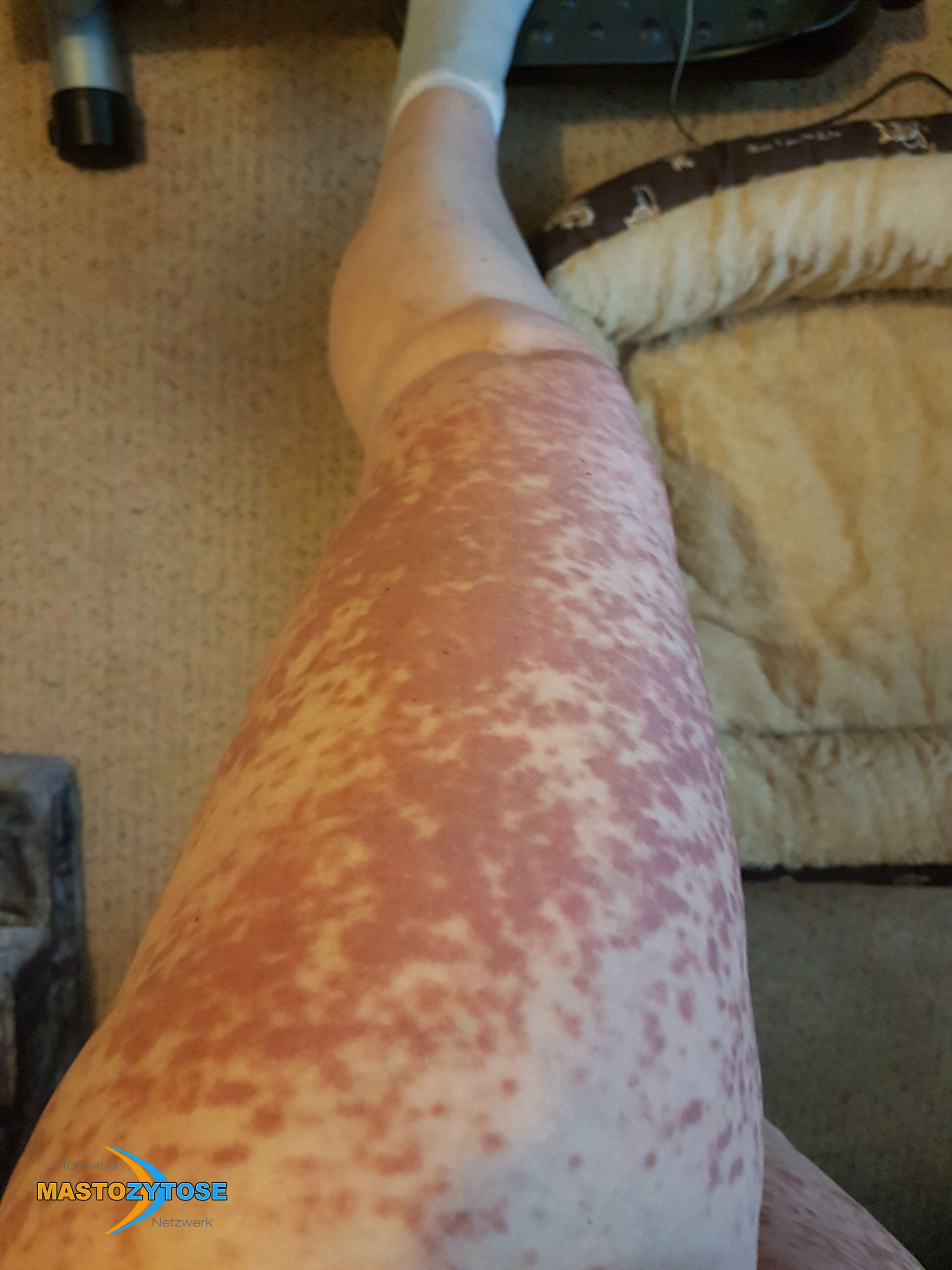
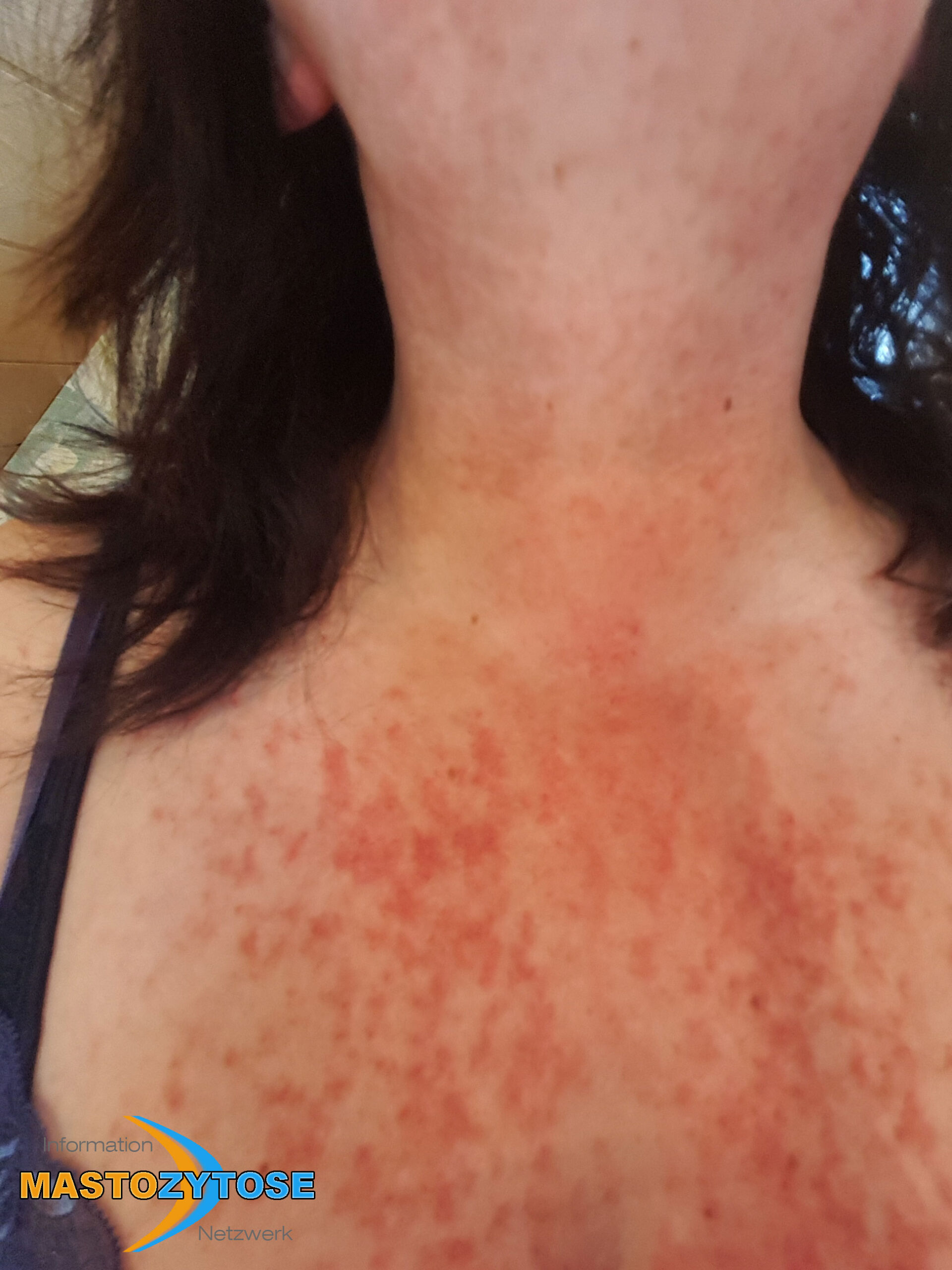
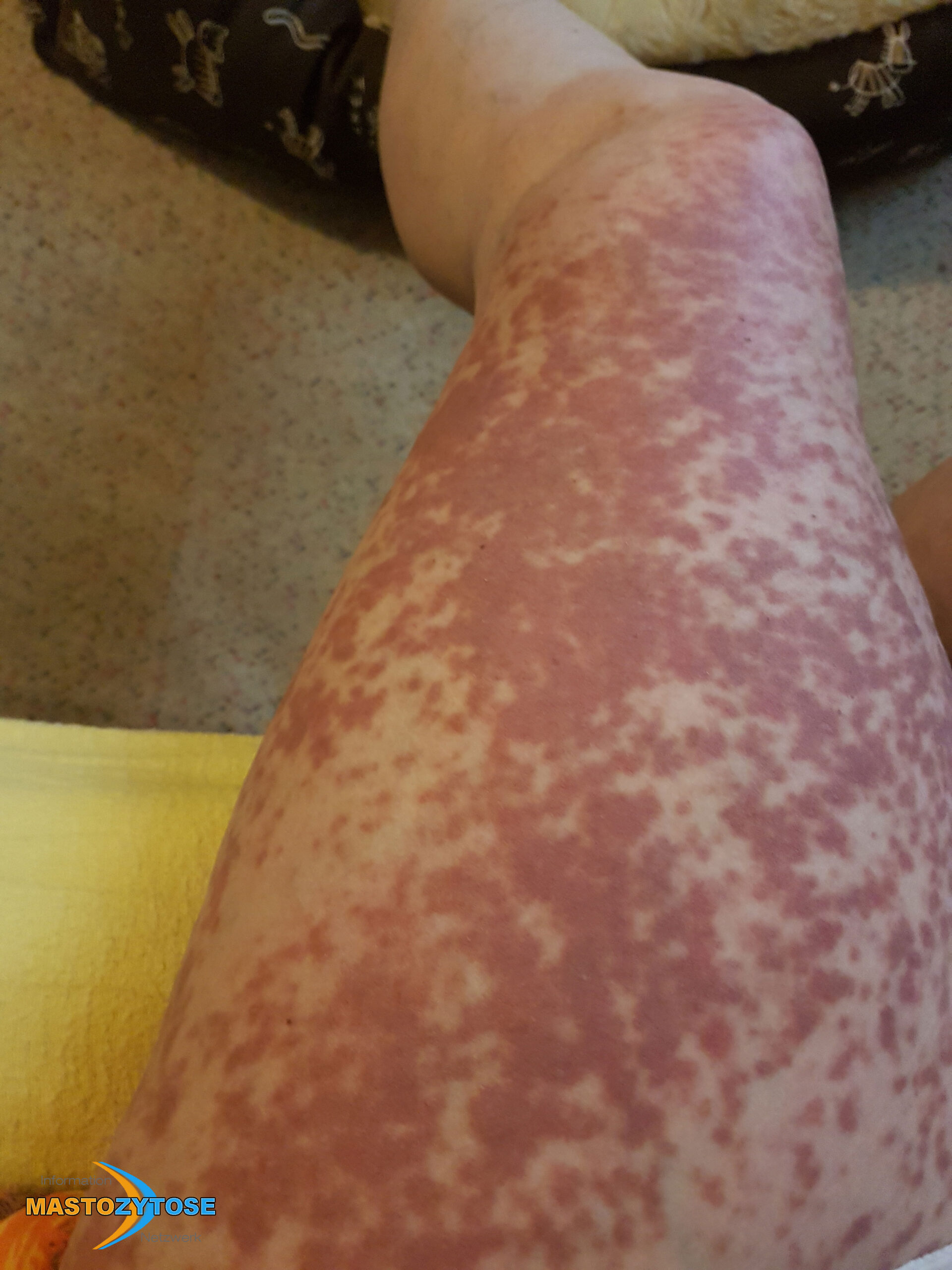

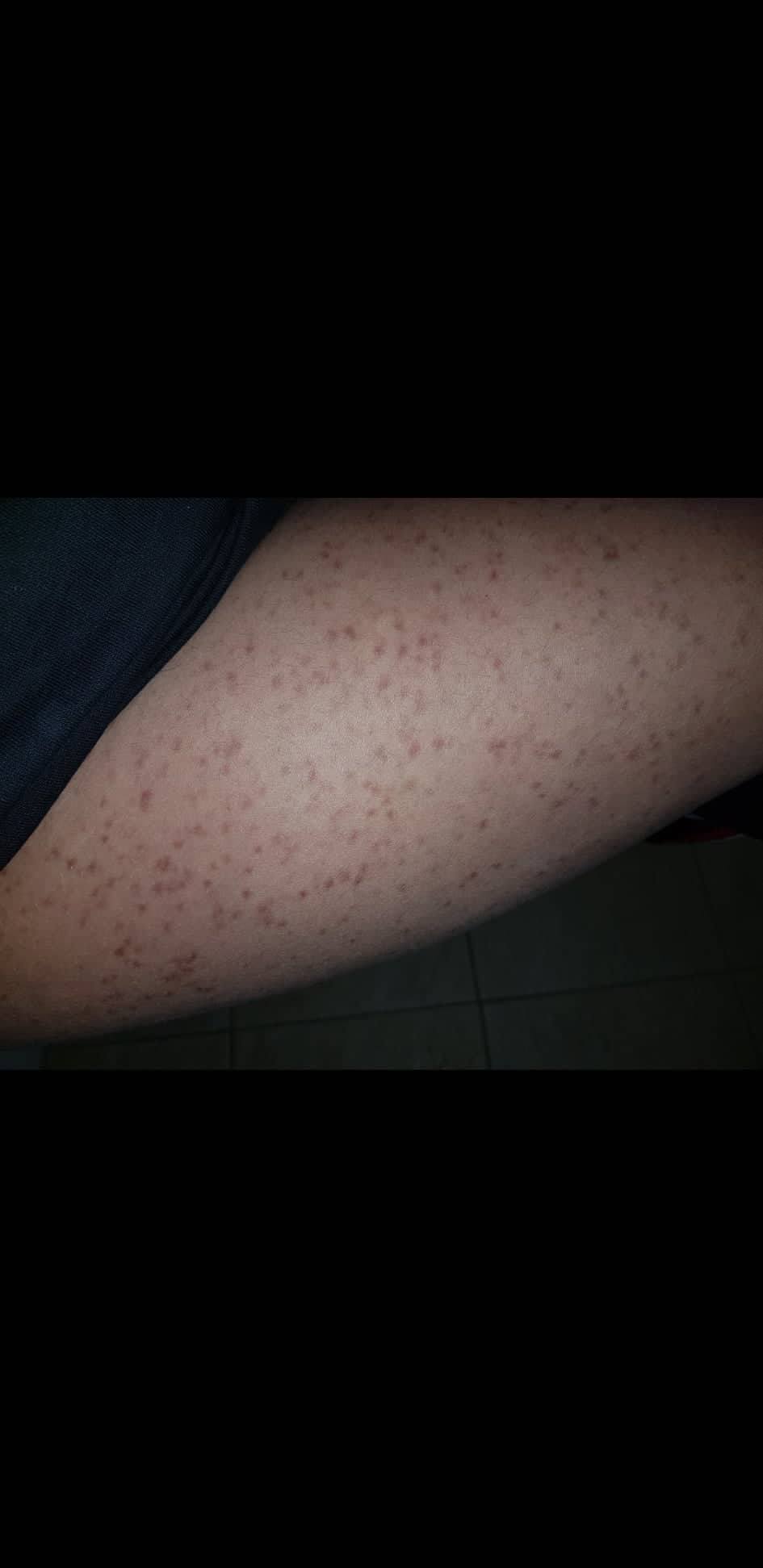
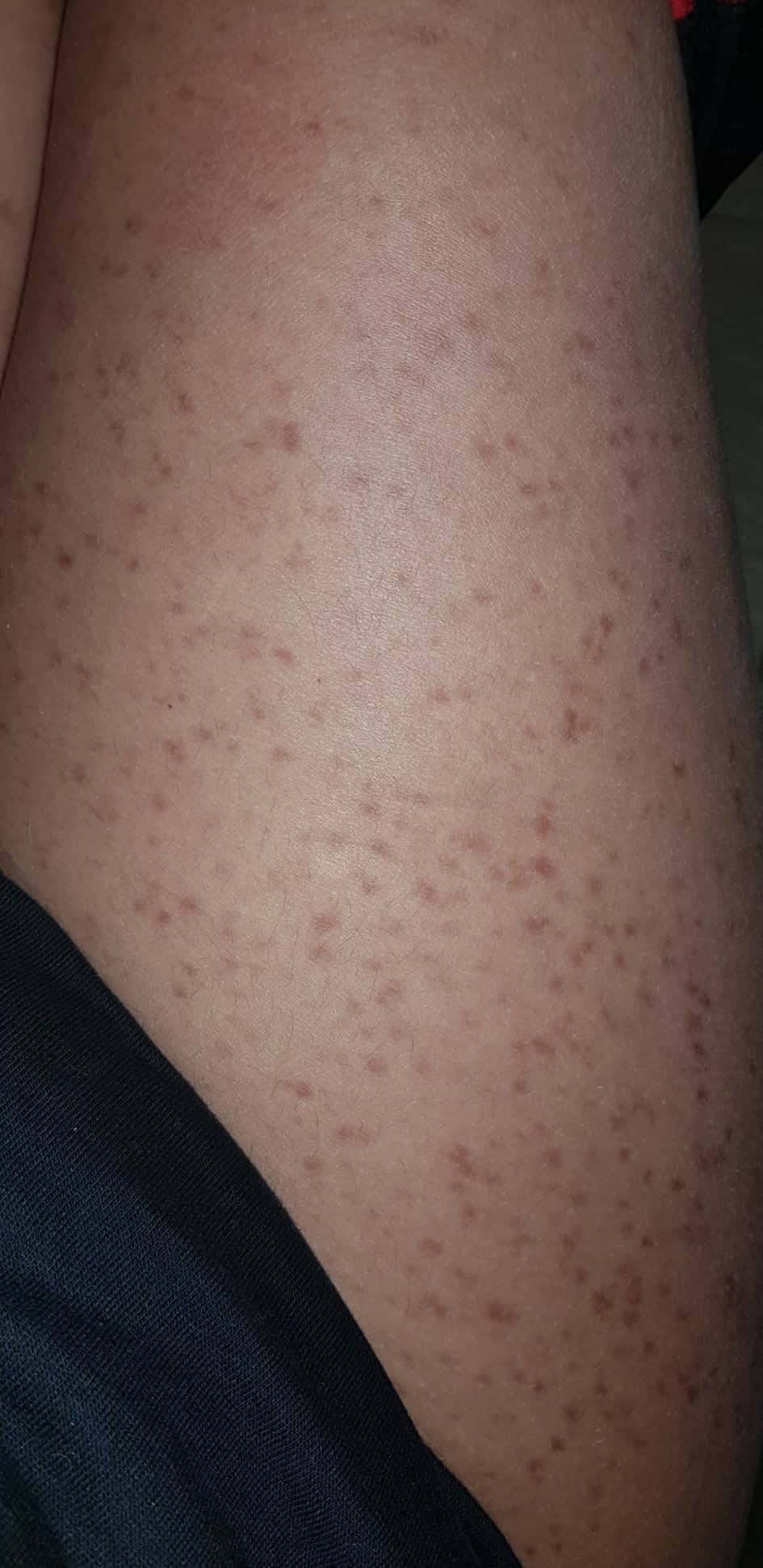
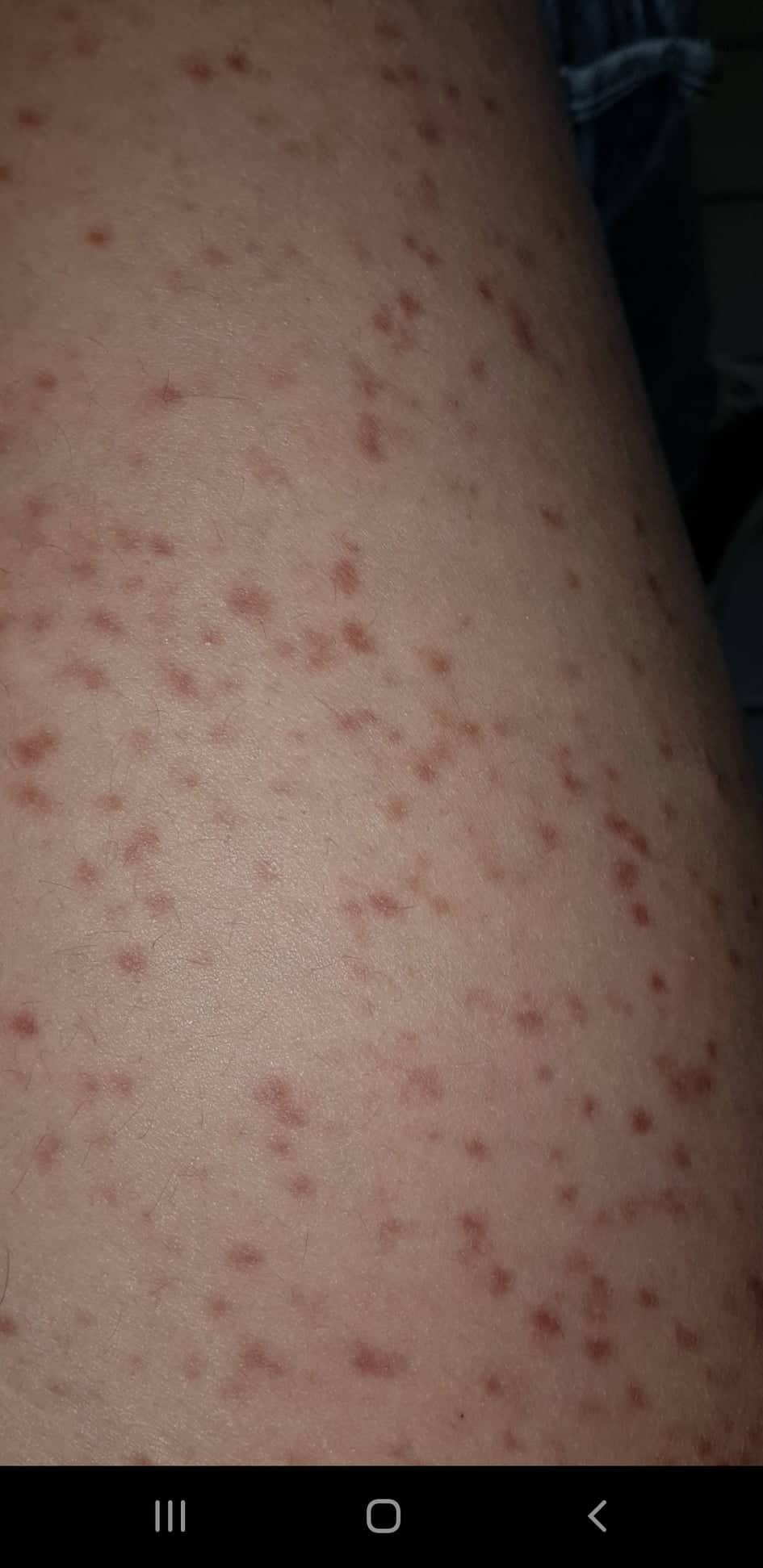
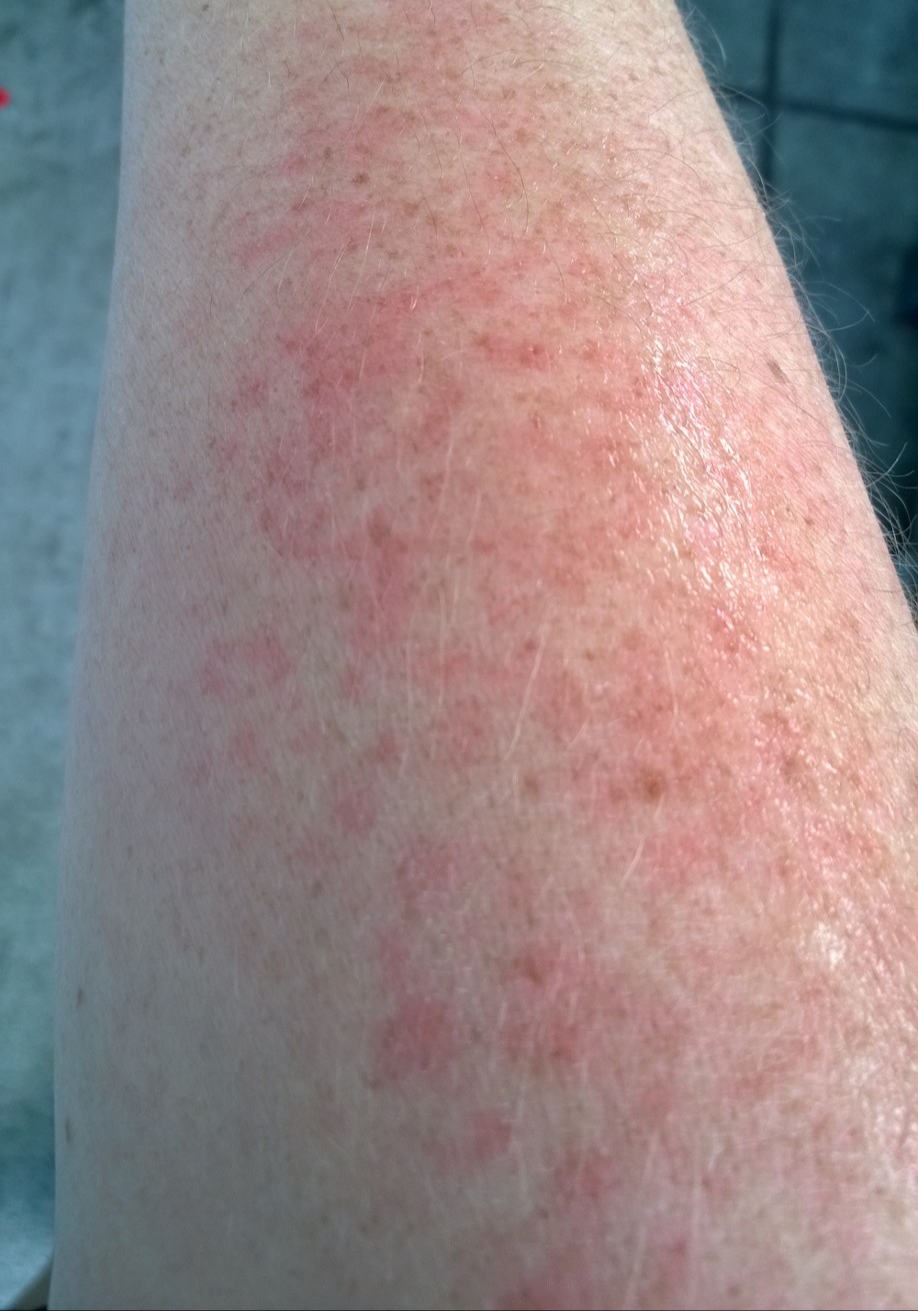
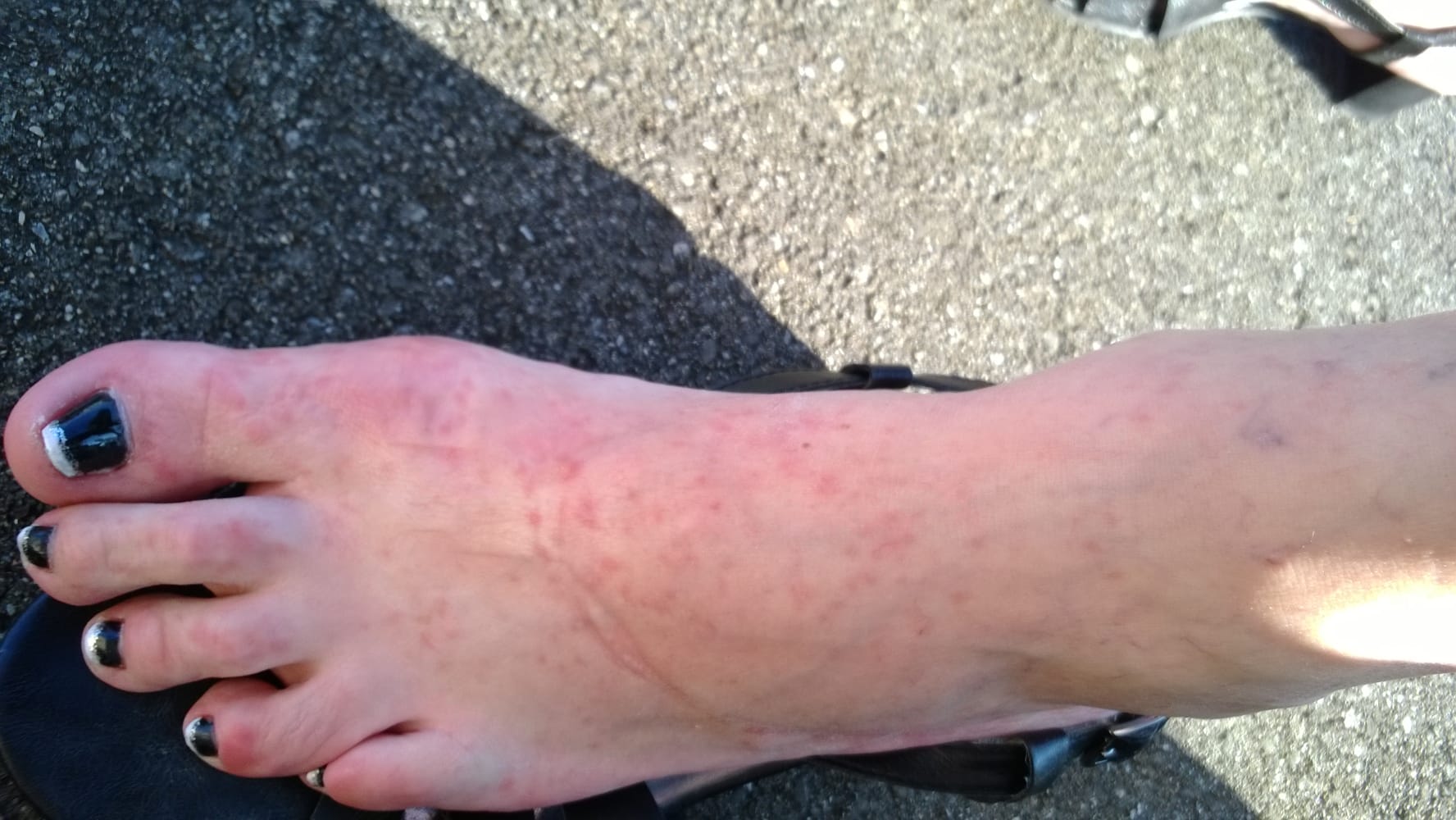
Symptome im Körperinneren
Treten Beschwerden woanders als auf der Haut auf, handelt es sich in der Regel um die systemische Mastozytose. Aber auch bei der Hautmastozytose können Erwachsene Symptome an anderen Stellen des Körpers bekommen.
Bei der Mastozytose entstehen die Symptome durch die Botenstoff-Freisetzung der Mastzellen oder durch die hohe Anzahl der Mastzellen selbst (mehr Informationen dazu finden Sie im Artikel Ursachen). Bei der fortgeschrittenen Mastozytose können Organe in ihrer Funktion bereits geschädigt sein.
Hier ein Überblick über die verschiedenen Anzeichen, die überall im Körper auftreten können:
Verdauungstrakt und innere Organe
- Sodbrennen
- Übelkeit
- Erbrechen
- Durchfall
- Bauchkrämpfe
- Magengeschwüre
- Lebervergrößerung
- Milzvergrößerung
- Gewichtsverlust
- Ansammlung von Wasser in der Bauchhöhle (Aszites)
Atemwege, Schleimhäute, Blut
- Hustenreiz
- Schwellung des Nasen-Rachenraumes
- Schwellung des Kehlkopfes
- Stimmstörung
- vergrößerte Lymphknoten
- Veränderung des Blutbildes
Herz-Kreislaufsystem
- Schwindel
- Müdigkeit
- Schwächegefühl
- Herzrasen
- anfallsartiger Blutdruckabfall
- allergischer Schock
Nervensystem
- Gedächtnis- und Konzentrationsstörungen
- Depression
- Kopfschmerzen
- Schlafstörungen
- allgemeine Schwäche, Abgeschlagenheit, Fatigue
- Reizbarkeit
Knochen und Muskeln
- diffuse Knochenschmerzen
- Muskelschmerzen
- Knochenschwund
- Knochenbrüche
- Osteolysen (Knochendefekt durch Abbau der Knochen)
Genitalbereich
- Schmerzen oder Brennen in der Harnblase
- Harndrang
- Menstruationsbeschwerden
Wie ist die Lebensqualität bei Mastozytose?
Die genannten Symptome können in unterschiedlichen Schweregraden vorkommen – von mild bis lebensbedrohlich.
In der Regel ist die Lebenserwartung bei der (indolenten) systemischen Mastozytose nur wenig oder gar nicht eingeschränkt, aber die Lebensqualität kann darunter leiden. Betroffene haben beispielsweise starke Lebensmittelunverträglichkeiten, besonders histaminhaltige Speisen und Getränke wie Käse, Rotwein, Nüsse oder Schokolade können Beschwerden hervorrufen. Außerdem können Schmerzen in verschiedenen Teilen des Körpers auftreten. Manchmal haben Betroffene so starke Magen-Darm-Probleme, dass sie sich kaum noch in die Öffentlichkeit trauen oder beruflich eingeschränkt sind. Bei 1 bis 3 von 10 Personen mit systemischer Mastozytose nehmen die Knochen an Dichte und Festigkeit ab und können leicht brechen. Das nennt sich Osteoporose oder in der Vorstufe Osteopenie. Die Mehrzahl aller Betroffenen mit einer (indolenten) systemischen Form lernt mit ihren Symptomen gut umzugehen.
Bei der fortgeschrittenen systemischen Mastozytose sind meist innere Organe wie Leber oder Milz vergrößert und funktionieren nicht mehr richtig. Leider kann bei Menschen mit einer fortgeschrittenen Mastozytose die Lebenserwartung verkürzt sein. Auch die Lebensqualität ist je nach Schwere der Symptome häufig eingeschränkt. Die vergrößerten Organe können beispielsweise Schmerzen auslösen. Zudem kann es vorkommen, dass zu wenig Blut gebildet wird und die Betroffenen müde und abgeschlagen sind. In manchen Fällen kann im Magen-Darm-Trakt die Nahrung nicht mehr richtig aufgenommen werden und es kommt zu Mangelernährung und Gewichtsverlust. Bei einer fortgeschrittenen Form können sich die Mastzellen so stark krankhaft verändern (entarten), dass Betroffene Blutkrebs (Mastzell-Leukämie) entwickeln. Mehr zum Verlauf der Erkrankung finden sie im Artikel „Ursachen“.
Was sind Trigger bei Mastozytose?
Mastzellen schütten durch bestimmte Auslöser verschiedene Botenstoffe aus, der bekannteste ist Histamin. Diese Stoffe verursachen die Symptome der Mastozytose. Die Mastzellen setzen die Botenstoffe frei, wenn sie in Kontakt mit bestimmten Reizen kommen, auch Trigger genannt. Typische Trigger sind:
- Infekte
- Stress, Aufregung, starke Emotionen
- Insektenstiche, Quallengifte, Schimmel, Umweltgifte
- körperliche Anstrengung, Schlafmangel
- plötzliche Temperaturwechsel
- Nahrungsmittel und Zusatzstoffe
- Alkohol
- Düfte (Parfum), Gerüche
- Medikamente, wie beispielsweise Schmerzmittel, Morphin, Röntgenkontrastmittel, Narkosemedikamente (was speziell vor Operationen zu beachten ist, haben wir in einem OP-Flyer zusammengefasst)
- Druck und Reibung auf der Haut (beispielsweise Massagen)
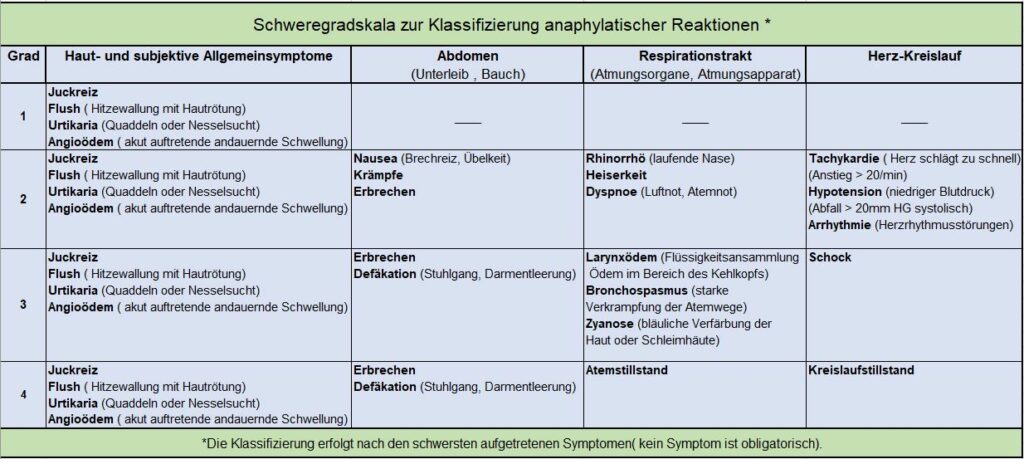
Notfall: Was ist ein anaphylaktischer Schock bei Mastozytose?
Wenn viele Mastzellen gleichzeitig ihre Botenstoffe ausschütten, kann dies zu einem lebensbedrohlichen Schock führen. Dabei treten neben den bekannten Symptomen auch Atemnot, Herzrasen, Schwindel und Bewusstlosigkeit auf.
Bei Anzeichen wie Atemnot oder Kreislaufzusammenbruch sollte sofort ein Krankenwagen gerufen werden!
Menschen mit Mastozytose sollten immer einen Notfall-Ausweis und ein Notfall-Set mit Medikamenten dabeihaben, die von der behandelnden Ärztin oder dem Arzt zusammengestellt werden. Fachleute empfehlen, die Familie und das soziale Umfeld über die Erkrankung zu informieren, damit diese wissen, was im Notfall zu tun ist.
Es ist zudem wichtig, dass Betroffene ihre Trigger kennen und möglichst vermeiden.
Mastozytose Awareness Teaser
20.10 ist der Internationale Tag der #Mastozytose und der Mastzellerkrankung: Dieser Tag soll uns alle daran erinnern, dass es diese Erkrankungen gibt! Dies ist ein kleiner Trailer, um auf die Besonderheit der Krankheit Mastozytose und auf den Awareness-Tag hinzuweisen.
Zusammenfassung Symptome und Formen der Mastozytose
Mastozytose ist eine Krankheit mit vielen Gesichtern – sie kann in unterschiedlichen Formen auftreten und zahlreiche Beschwerden verursachen, die individuell sehr verschieden sein können. Welche Triggerfaktoren Symptome auslösen, kann sich ebenfalls von Person zu Person unterscheiden. Wie sehr eine Mastozytose die Gesundheit und den Alltag von Betroffenen einschränkt ist daher sehr variabel – manche Menschen haben kaum Beschwerden und fühlen sich nicht beeinträchtigt, anderen Menschen sind schwer krank und können Freizeitaktivitäten und Beruf nicht mehr ausüben. Daher spielen die genaue Diagnostik der Mastozytose-Form und die individuell abgestimmte Behandlung eine wichtige Rolle.
Weiterlesen
Bundesärztekammer: Patienteninformation Mastozytose
Interdisziplinäres Mastozytose Centrum Charité: Über Mastozytose – Formen, Krankheitsbild und Behandlungsmöglichkeiten
Patienten-Information des ÄZQ: Mastozytose – welche Formen gibt es?
Quellenangaben
Deutsche Gesellschaft für Allergologie und klinische Immunologie (DGAKI) et al. Leitlinie Anaphylaxie (S2k-Leitlinie). AWMF-Registernr.: 061-025. 2021.
Bundesärztekammer. 2018. Patienteninformation Mastozytose. Abruf am 21.07.2022 von https://www.bundesaerztekammer.de/fileadmin/user_upload/_old-files/downloads/pdf-Ordner/Patienteninformationen/mastozytose.pdf
Deutsche Gesellschaft für Hämatologie und Medizinische Onkologie e.V. (DGHO). Systemische Mastozytose. Abruf am 21.07.2022 von https://www.onkopedia.com/de/onkopedia/guidelines/mastozytose-systemische/@@guideline/html/index.html
Interdisziplinäres Mastozytose Centrum Charité. Über Mastozytose – Formen, Krankheitsbild und Behandlungsmöglichkeiten. Abruf am 21.07.2022 von https://imcc.charite.de/fuer_patienten/
Hans-Peter Horny, Karl Sotlar, Peter Valent und Karin Hartmann. 2008. Deutsches Ärzteblatt International; 105(40): 686–92. Abruf am 26.07.2022 von https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2696962/pdf/Dtsch_Arztebl_Int-105-0686.pdf
Nicola Wagner und Karin Hartmann. 2011. Mastozytose – Diagnostisches und therapeutisches Management. Akt Dermatol 2011; 37: 419–427. Thieme connect. Abruf am 19.8.2022 von https://www.thieme-connect.com/products/ejournals/pdf/10.1055/s-0030-1256887.pdf